Decoding the effects of hydrophobic mutations in a long intrinsically disordered protein
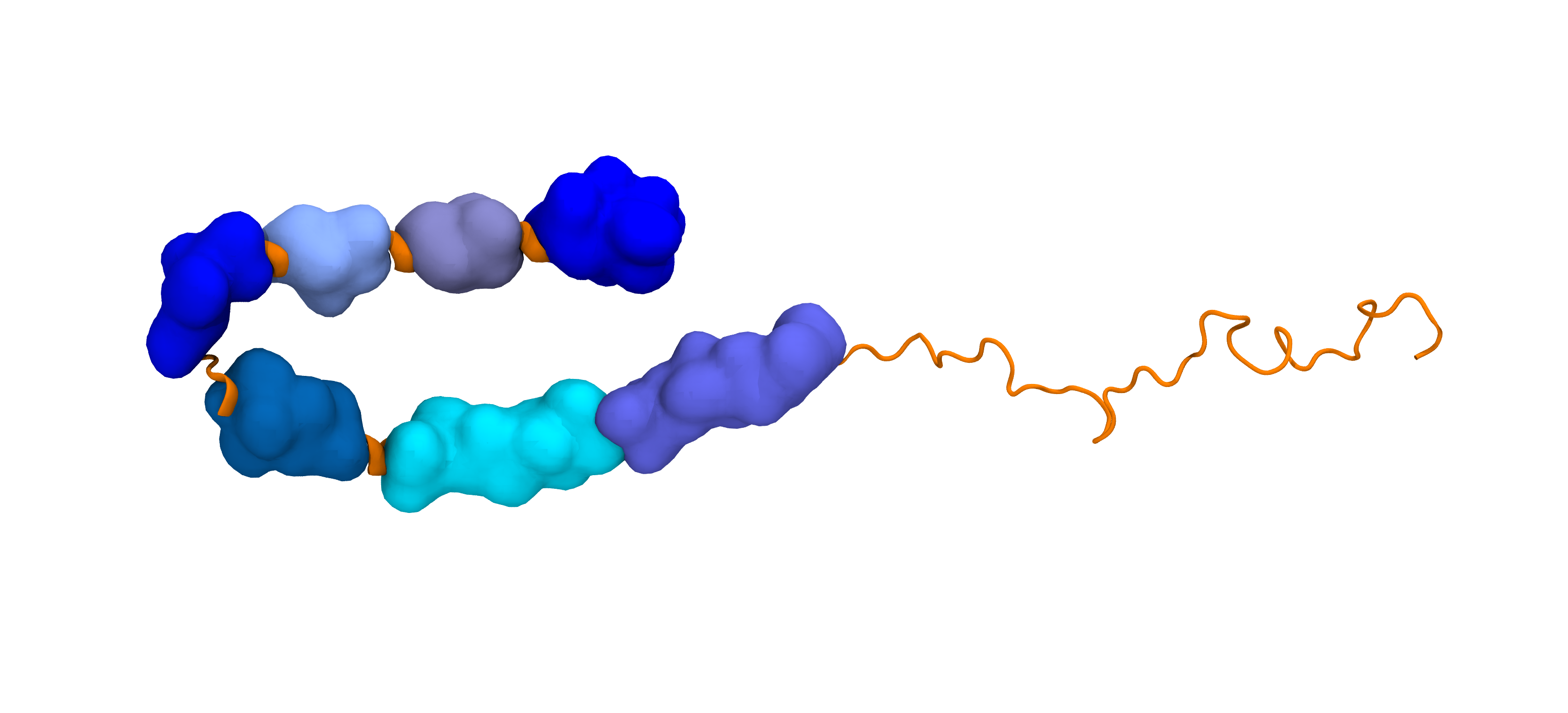
A naturally occurring Val66Met mutation near the center of the intrinsically disordered Brain-Derived Neurotrophic Factor (BDNF) prodomain sequence induces significant changes at the termini, altering protein function. This project addresses the question: "How can subtle residue changes far from the termini influence interactions between them?" To answer this question, we are developing a systematic approach to cluster the conformational ensemble based on contacts between contiguous hydrophobic regions, or “blobs.” Above, a BDNF prodomain variant is shown with QuickSurf representations of blobs, highlighting the blob containing the mutation at site 66 in pink.
Determining the effects of local sequence on Met-Met interactions in Intrinsically Disordered Proteins
While many proteins fold and have well-defined structures, many other proteins (and parts of proteins) do not. These are konwn as Intrinsically Disordered Proteins (IDPs). In the absence of tertiary structure, it is a major challenge to identify residue-residue interactions and overall confromational trends. This project involves using genomic data and simulations to elucidate patterns involving both of these in IDPs. Our lab developed the blobulator, an amino acid sequence-based edge detection tool - which can be found here.
Computing absolute binding affinities by Streamlined Alchemical Free Energy Perturbation
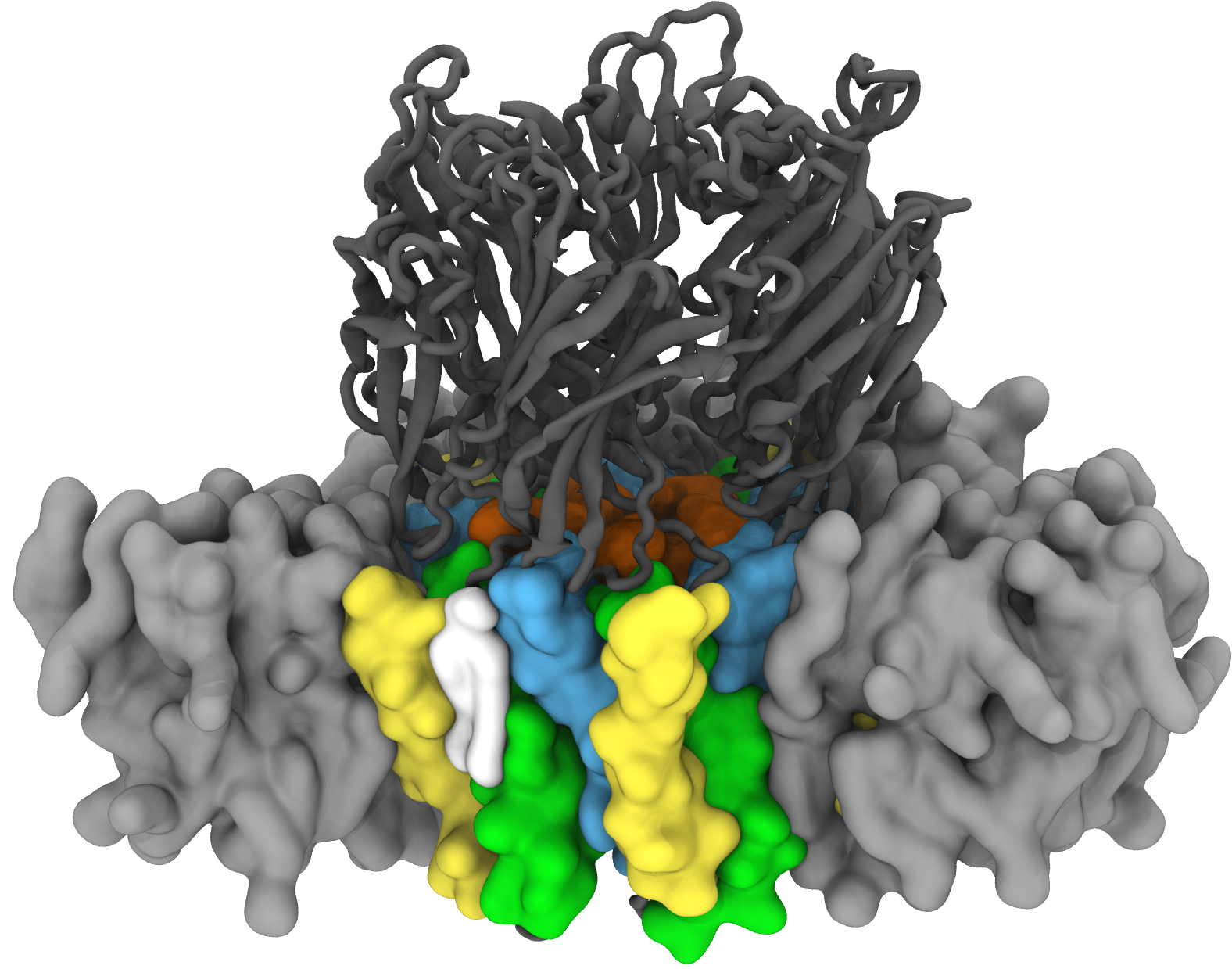
Not all cell membranes are identical; they’re made up of a wide range of lipids which can affect critical cell functions like metabolism or nerve signaling. We are trying to answer the question of “What is the probability that lipid X affects the function of membrane protein Y?” We get at that question by computing free energies of binding affinities using the SAFEP framework. In this framework we use molecular dynamics simulations to compare the bound and unbound states of the lipid and protein. Here we see a lipid specifically and tightly bound to the Erwinia ligand gated ion channel, a model for a large family of neuronal proteins, modulating the protein's activity. From our MD simulations, we can determine the probabilities of binding and so compare the effects of different lipids.
Asymmetric Membrane Curvature in SARS-CoV-2 E protein
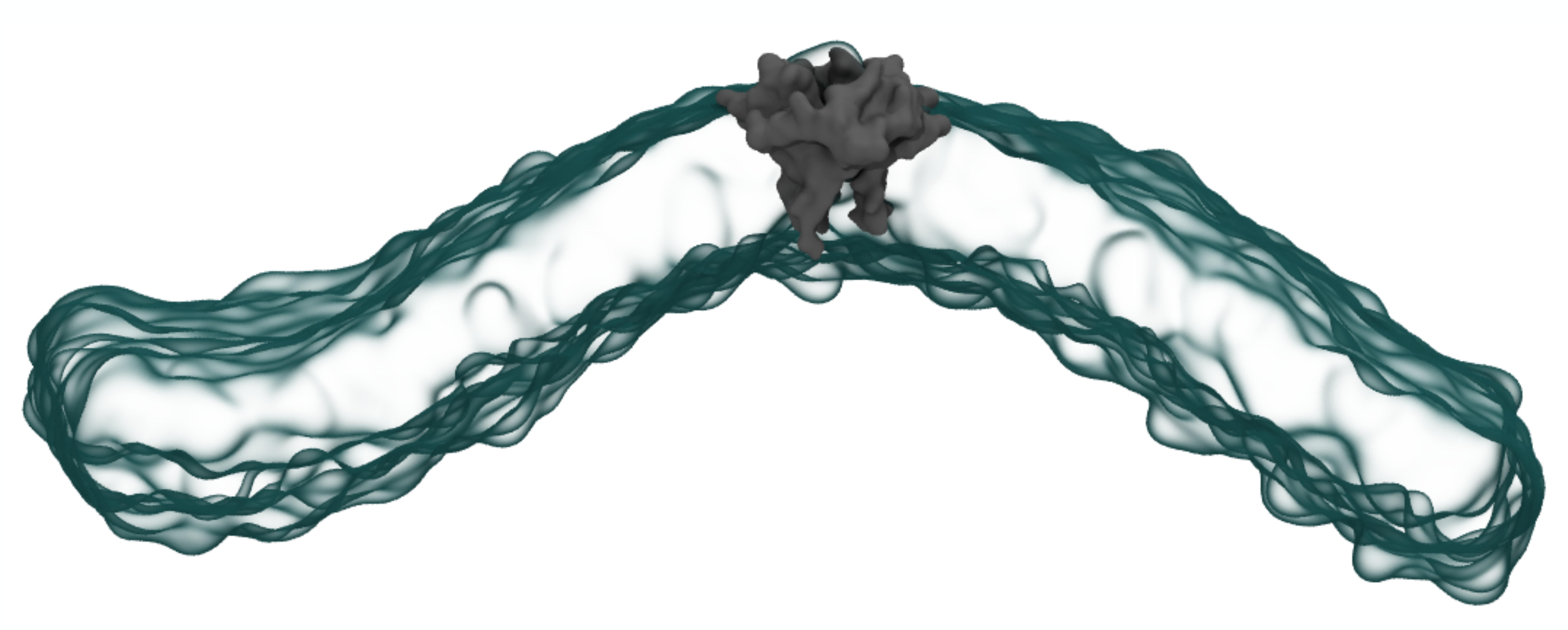
When the virus that causes Covid-19 replicates inside your cells, the Envelope protein (E protein) wraps the baby virion in a blanket of your own organellar membrane material. This helps the new virion escape detection by your immune system and allows it to escape its host cell, continuing the Covid-19 infection cycle. It is not known how this small, pentameric protein could induce such extreme curvature in a membrane surface, but that is what my research seeks to uncover. To understand the relationship between E protein and membrane curvature, we developed a toolkit for analysis of membrane disruption by proteins and other inclusions, called nougat. Shown above is a snapshot of the SARS-CoV-2 Envelope (E) protein (grey) bending a 100% POPC membrane (cyan) in coarse-grain molecular dynamics simulation.
The role of membrane composition on the aggregation and stability of nanoparticles
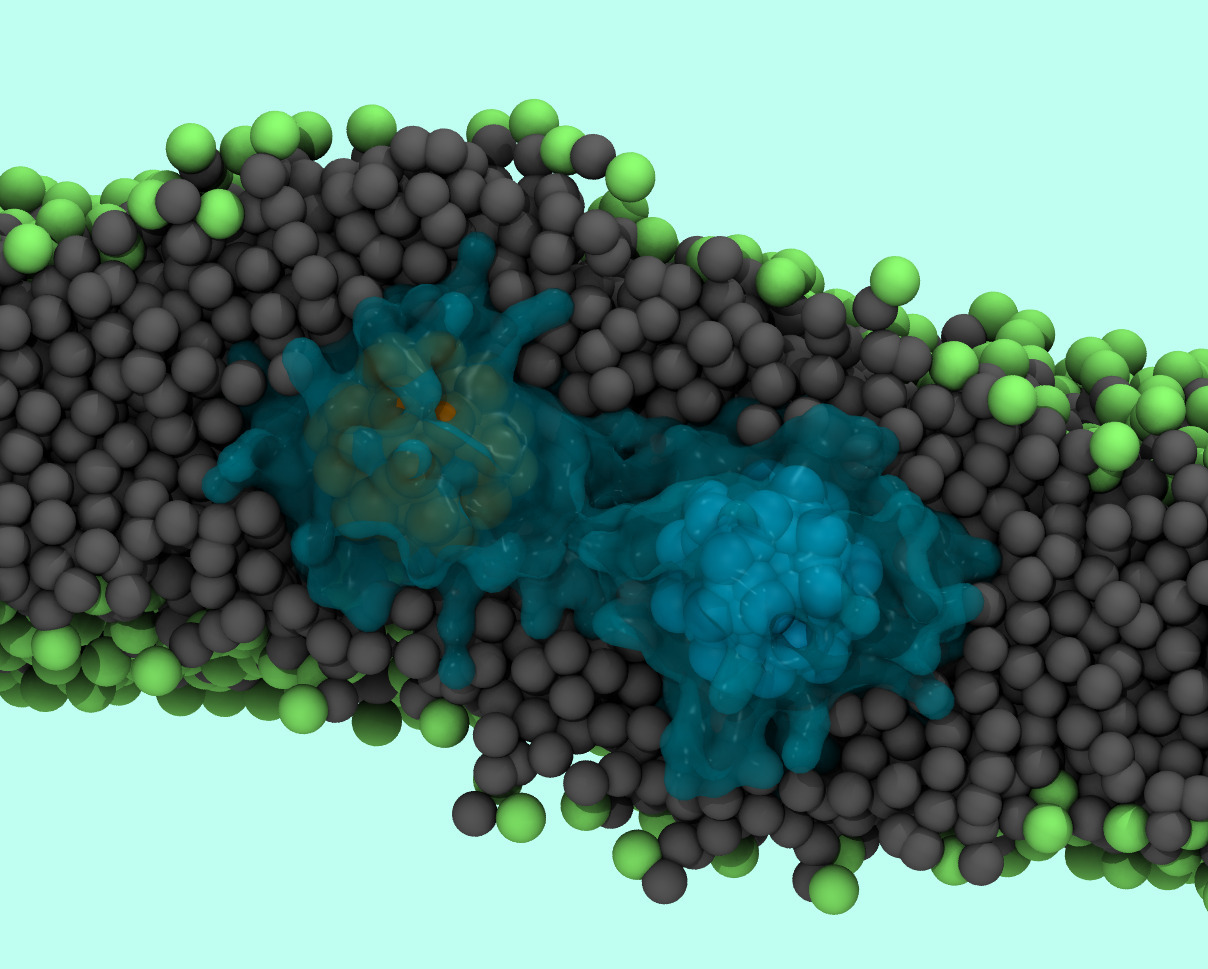
Targeted drug delivery is a rapidly expanding field, with promising technologies to control the spatial-temporal release of drugs. Some of the most promising delivery vehicles are polymer and lipid vesicles hybridized with metallic nanoparticles. Our primary focus is understanding nanoparticle-membrane interactions and molecular mechanisms underlying nanoparticle aggregation and lipid disruption around nanoparticle inclusions. We use molecular dynamics simulations to understand these interactions at the molecular level. Shown above is an image of two aggregated dodecanethiol coated charged nanoparticles in a POPC membrane. Nanoparticles are represented in ochre and cyan, ligands in blue, lipid tails in grey and lipid head groups in green.
Investigating the role of ceramides in regulating C. cresentus antibiotic sensitivity
Unlike many gram negative bacteria, the Caulobacter Crescents (C.cresentus) outer membrane contains sphingolipids, lipids with a sphingosine backbone. Under low phosphate conditions, C.cresentus increases sphingolipid synthesizes; furthermore, it has been shown that disruption of sphingolipid synthesis leads to changes in antibiotic sensitivity. Our primary research goal is to understand the mechanism of sphingolipid driven antibiotic sensitivity at the molecular level. Red: O-antigen, Yellow + Cyan: Lipid A, Orange + Grey: Anionic Sphingolipid, Green + Iceblue: POPG.